Multisample analysis
Contents
Multisample analysis#
Setup#
import missionbio.mosaic as ms
from IPython.core.display import display, HTML
/var/folders/4g/9gyytq3n1m584g8yr5jxch480000gn/T/ipykernel_86479/1021175102.py:2: DeprecationWarning: Importing display from IPython.core.display is deprecated since IPython 7.14, please import from IPython display
from IPython.core.display import display, HTML
# Load a multi-sample file
# By default, a multi-sample h5 file loads as a SampleGroup object
group = ms.load_example_dataset("Multisample PBMC")
Loading, <_io.BytesIO object at 0x7fa939143b30>
Loaded in 2.5s.
# To analyze one sample at a time access, them using the samples attribute
sample = group.samples[1]
# These are the samples in the h5 file
[s.name for s in group.samples]
['Sample 1', 'Sample 2']
Applying functions#
# To apply a function across all samples use the `apply` method on the SampleGroup
# It returns a list of returned objects for each sample
def filt(sample):
filt_vars = sample.dna.filter_variants()
return filt_vars
filtered_variants = group.apply(filt)
filtered_variants
[array(['chr2:198266943:C/T', 'chr2:198267770:G/GAA', 'chr4:55599436:T/C',
'chr5:170837457:A/G', 'chr7:140449071:C/G', 'chr7:148504716:AG/A',
'chr7:148504854:A/AGACTT', 'chr7:148508833:A/G',
'chr7:148543525:A/G', 'chr7:148543583:G/C', 'chr7:148543693:TA/T',
'chr11:32417945:T/C', 'chr11:119148573:G/T', 'chr13:28602292:T/C',
'chr17:7578115:T/C', 'chr17:29559932:C/A', 'chr20:31024028:G/A',
'chrX:39933339:A/G', 'chrX:44833841:C/A', 'chrX:133547814:T/C'],
dtype='<U23'),
array(['chr2:198266943:C/T', 'chr2:198267770:G/GAA', 'chr4:55599436:T/C',
'chr5:170837457:A/G', 'chr7:140449071:C/G', 'chr7:148504716:AG/A',
'chr7:148504854:A/AGACTT', 'chr7:148508833:A/G',
'chr7:148543525:A/G', 'chr7:148543583:G/C', 'chr7:148543693:TA/T',
'chr11:32417945:T/C', 'chr11:119148573:G/T', 'chr13:28602292:T/C',
'chr17:7578115:T/C', 'chr17:7579801:G/C', 'chr17:29559932:C/A',
'chr20:31024028:G/A', 'chrX:39933339:A/G', 'chrX:44833841:C/A',
'chrX:133547814:T/C'], dtype='<U23')]
# Subset the same variants in all dna assays
# It is important to maintain the same variants across all dna assays
og_num_vars = [s.dna.shape[1] for s in group.samples]
var_union = list(set().union(*filtered_variants))
for sample in group:
sample.dna = sample.dna[:, var_union] # Subsets all samples with the same variants
new_num_vars = [s.dna.shape[1] for s in group.samples]
print(og_num_vars, new_num_vars) # Thee old and new number of variants for each sample in the group
[21, 21] [21, 21]
# The functions applied on each sample can be more complex - like this assignment and relabeling method
# Note the original labels can be uncoordinated across samples in the group
# The labels are changed to ensure that each label is for the same clone
variants_of_interest = ['chr7:148508833:A/G', 'chr17:29559932:C/A', 'chr4:55599436:T/C']
def cluster(sample):
clone_table = sample.dna.group_by_genotype(variants_of_interest)
# Rename labels so that each sample has the same labels
# Here the signature of each variant is used to rename the labels
df = sample.dna.signature("NGT").loc[:, variants_of_interest]
names = df.apply(lambda vs: "-".join([str(int(v)) for v in vs]), axis=1)
label_map = {i: n for i, n in names.items()}
del label_map["missing"]
del label_map["small"]
sample.dna.rename_labels(label_map)
clone_table = clone_table.rename(index=label_map)
return clone_table # Return the clone table
tables = group.apply(cluster)
[display(HTML(t.to_html())) for t in tables] # The clone tables for each sample
| clone | 1 | 2 | 4 | 3 | 5 | 6 | 7 | Missing GT clones (4) | Small subclones (12) | ADO clones (0) |
|---|---|---|---|---|---|---|---|---|---|---|
| chr7:148508833:A/G | Het (50.2%) | Het (50.34%) | WT (2.14%) | WT (3.83%) | Het (49.45%) | Het (35.02%) | Hom (97.55%) | Missing in 0.00% of cells | WT (32.3%) | NaN |
| chr17:29559932:C/A | Het (49.37%) | Hom (97.39%) | Het (51.98%) | Het (50.86%) | WT (2.82%) | Het (49.73%) | Het (47.63%) | Missing in 0.10% of cells | Het (49.89%) | NaN |
| chr4:55599436:T/C | Hom (99.81%) | Hom (99.75%) | Het (53.44%) | Hom (99.46%) | Hom (99.74%) | Het (73.29%) | Hom (99.93%) | Missing in 0.67% of cells | Hom (70.91%) | NaN |
| Total Cell Number | 1377 (70.87%) | 89 (4.58%) | 87 (4.48%) | 87 (4.48%) | 86 (4.43%) | 79 (4.07%) | 67 (3.45%) | 15 (0.77%) | 56 (2.88%) | NaN |
| Sample 1 Cell Number | 1377 (70.87%) | 89 (4.58%) | 87 (4.48%) | 87 (4.48%) | 86 (4.43%) | 79 (4.07%) | 67 (3.45%) | 15 (0.77%) | 56 (2.88%) | NaN |
| Parents | [6] | [1] | NaN | [1, 4] | [1] | NaN | [1] | NaN | NaN | NaN |
| Sisters | [small] | [5] | NaN | [7, small] | [2] | NaN | [3] | NaN | NaN | NaN |
| ADO score | 0.0 | 0.965058 | 0 | 0.934829 | 0.957791 | 0 | 0.961455 | NaN | NaN | NaN |
| clone | 1 | 2 | 3 | 4 | 5 | 6 | 7 | Missing GT clones (4) | Small subclones (7) | ADO clones (0) |
|---|---|---|---|---|---|---|---|---|---|---|
| chr7:148508833:A/G | WT (1.03%) | WT (0.82%) | WT (1.62%) | Het (29.15%) | WT (0.7%) | Het (49.14%) | WT (0.57%) | Missing in 0.00% of cells | WT (20.71%) | NaN |
| chr17:29559932:C/A | Het (49.45%) | Het (47.12%) | Het (46.38%) | Het (49.2%) | Hom (98.04%) | Het (49.25%) | WT (3.19%) | Missing in 0.06% of cells | Het (47.46%) | NaN |
| chr4:55599436:T/C | Het (50.56%) | WT (7.08%) | Hom (97.88%) | Het (70.28%) | Het (55.46%) | Hom (99.42%) | Het (55.66%) | Missing in 0.94% of cells | Hom (67.46%) | NaN |
| Total Cell Number | 1189 (65.76%) | 148 (8.19%) | 93 (5.14%) | 79 (4.37%) | 78 (4.31%) | 74 (4.09%) | 63 (3.48%) | 18 (1.0%) | 66 (3.65%) | NaN |
| Sample 2 Cell Number | 1189 (65.76%) | 148 (8.19%) | 93 (5.14%) | 79 (4.37%) | 78 (4.31%) | 74 (4.09%) | 63 (3.48%) | 18 (1.0%) | 66 (3.65%) | NaN |
| Parents | NaN | [1] | [1, 6] | NaN | [1] | NaN | [1] | NaN | NaN | NaN |
| Sisters | NaN | [3] | [2, small] | NaN | [7] | NaN | [5] | NaN | NaN | NaN |
| ADO score | 0 | 0.854166 | 0.999992 | 0 | 0.981959 | 0 | 0.959246 | NaN | NaN | NaN |
[None, None]
Drawing figures#
# Displaying the same plot across all the samples
var_order = group.samples[0].dna.clustered_ids("AF_MISSING")
for sample in group:
sample.dna.heatmap("AF_MISSING", features=var_order).show()
# Normalize CNV and look for patterns
for sample in group:
sample.protein.normalize_reads()
sample.heatmap(clusterby="dna", sortby="protein", drop="cnv", flatten=False).show()
variants_of_interest = ['chr17:29559932:C/A', 'chr4:55599436:T/C', 'chr7:148508833:A/G']
proteins_of_interest = ['CD19', "CD34", "CD30"]
clones_of_interest = ["0-1-1", "1-1-2"]
for sample in group:
s = sample[sample.dna.barcodes(clones_of_interest)]
s.dna = s.dna[:, variants_of_interest]
s.protein = s.protein[:, proteins_of_interest]
s.clone_vs_analyte("protein")
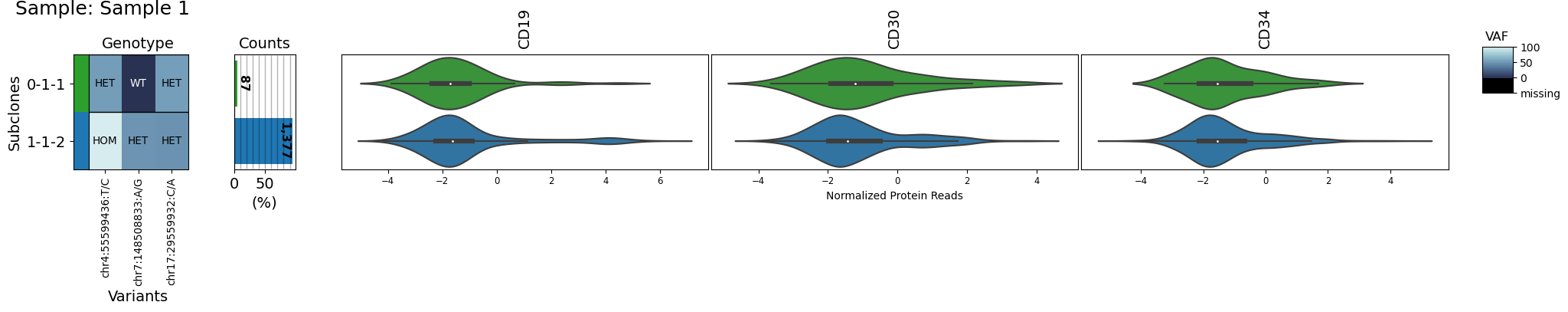
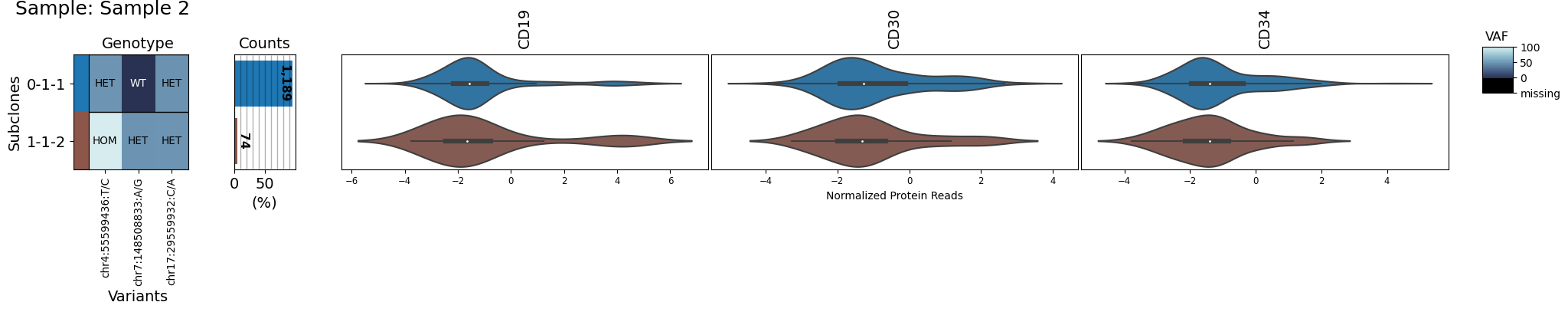
Multisample plots#
# Draw a fishplot for the dna labels
# From the proportions in the heatmaps - The two clones of interest are 0-1-1 and 2-20
group.fishplot(
"dna",
sample_order=["Sample 1", "Sample 2"],
labels=["0-1-1", "1-1-2"],
parents=[None, None]
)
# Draw a barplot for the dna labels
group.barplot(
"dna",
sample_order=["Sample 1", "Sample 2"],
label_order=["0-1-1", "1-1-2"],
percentage=True
)
/Users/casp/Documents/code/mosaic/src/missionbio/mosaic/samplegroup.py:334: FutureWarning:
Support for multi-dimensional indexing (e.g. `obj[:, None]`) is deprecated and will be removed in a future version. Convert to a numpy array before indexing instead.