Filtering barcodes and ids
Contents
Filtering barcodes and ids#
Objective
This vignette describes how data can be filtered in Mosaic.
filtering barcodes and filtering ids both are covered.
The h5 file used in this notebook can be found here
# Import mosaic and load the data
import missionbio.mosaic as ms
sample = ms.load_example_dataset("3 cell mix")
Loading, <_io.BytesIO object at 0x7fbd9990e900>
Loaded in 0.4s.
This is an analyzed h5 file. Hence the clones and clusters
are already labeled. It contains three cell lines KG-1,
Tom-1, and Jurkat. It also contains doublets of each pair
as seen in the heatmap.
sample.dna.heatmap('NGT')
Filtering assays#
Each assay and sample in Mosaic can be filtered
using the slice notation in Python. There is also
the drop function in an Assay object which can
be used to drop certain barcodes or ids.
In case any visualization is needed for a subset of
barcodes they can be dropped as follows.
Dropping barcodes and IDs#
This method is useful if certain barcodes or ids are to be dropped
# This shows all the labels present in assay
set(sample.dna.get_labels())
{'Jurkat', 'KG-1', 'Mixed', 'TOM-1'}
# Since this is an analyzed h5 file we can
# retrive all the barcodes labeled as Mixed
mixed_barcodes = sample.dna.barcodes('Mixed')
dna = sample.dna.drop(mixed_barcodes)
set(dna.get_labels())
{'Jurkat', 'KG-1', 'TOM-1'}
# Ids can be dropped in a similar fashion
# The id chosen was a poor quality variant in KG-1 as seen in the heatmap
dna = dna.drop(['chr2:25470426:C/T'])
# Once the cells and variants are dropped, we can make the plots as usual
# Here the mixed cells and that one id is dropped in the dna object.
dna.heatmap('NGT')
Selecting barcodes and IDs#
This is useful when only certain barcodes or IDs are to be selected
# This is an analyzed h5 file, hence Jurkat barcodes can be retrieved here
# In the slice notation, the `:` refers to all barcodes or all ids
# The first value is the subset of `barcodes` to be chosen,
# the second value is the subet of `ids` to be chosen.
jurkat_barcodes = sample.dna.barcodes('Jurkat')
dna = sample.dna[jurkat_barcodes, :]
set(dna.get_labels()) # This dna object only has Jurkat cells
{'Jurkat'}
# Let's say only two ids of interest are required for a plot
# We are reusing the dna object filtered earlier
id_of_interest = ['chr2:25458546:C/T', 'chr2:25469502:C/T']
dna = dna[:, id_of_interest]
dna.ids() # Has only the two ids selected
array(['chr2:25458546:C/T', 'chr2:25469502:C/T'], dtype=object)
# Once the assay has been filtered, it can be plotted
# We are now looking at the two selected variants for Jurkat cells only
dna.heatmap('NGT')
Filtering the entire sample#
If some barcodes are to be removed from all assays (DNA, Protein, CNV)
then, the sample level slice notation can be used.
# In this case, only a set of barcodes are required
# Choosing only two cell lines
cells = sample.dna.barcodes(['KG-1', 'TOM-1', 'Jurkat'])
filtered_sample = sample[cells]
# This is showing all the variants, but only for the three cell lines
filtered_sample.clone_vs_analyte(analyte='protein')
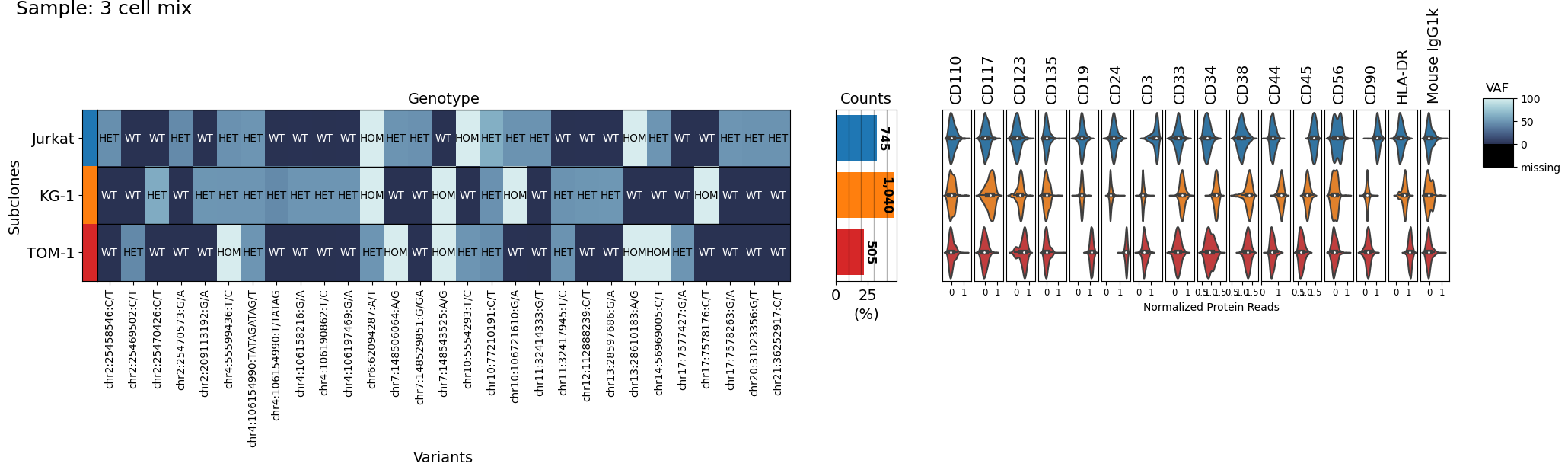
# To remove these variants the DNA object has to be filtered
# To remove some anitbodies, the protein object has to be filtered
variants = ['chr2:25458546:C/T', 'chr2:25469502:C/T']
filtered_sample.dna = filtered_sample.dna[:, variants] # Choosing all cells and two variants
abx = ['CD34', 'CD24', 'CD19', 'CD45', 'CD90']
filtered_sample.protein = filtered_sample.protein[:, abx] # Choosing all cells and four antibodies
# Now the plot only contains the cells and ids of interest
filtered_sample.clone_vs_analyte(analyte='protein')
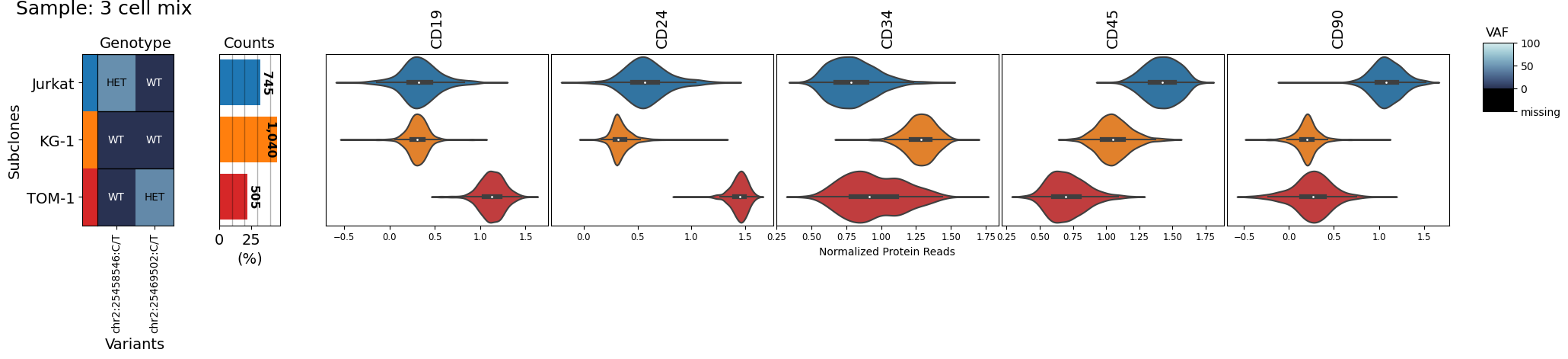
# The filtered sample object can be used for other plots as well
filtered_sample.heatmap(clusterby='dna', sortby='dna', drop='cnv', flatten=False)